
Panel Design: selecting fluorophores for spectral flow, part 1
- olivertburton

- Mar 4, 2024
- 5 min read
Once we've decided which markers to include in our panel, the next step is to begin selecting the fluorophores that we want to use in the panel and begin pairing those with markers. For spectral flow cytometry, I recommend starting by using the spectrum viewer tools we discussed last week to pick fluorophores that separate well from each other. First, we'll look at which types or combinations of fluorophores to avoid.
Today we're going to discuss an underappreciated problem that arises when similar fluorophores are used together in a panel: distortion of the negative (unstained) cells and loss of resolution. This loss of resolution can be thought of as uncertainty in assigning values, or as a twisting and stretching across the dimensions of the data, leading the negative to spread out. Negative distortion is independent of the spread we talked about here; for instance, we can have spread from fluorophores that share emission wavelengths without negative distortion if the fluorophores are excited by different wavelengths.
Let's look at a couple of examples so we can visualize the differences between spread and distortion. Spectral plots are from Cytek Cloud.
Example 1: BUV661 and APC


BUV661 and APC: separated well due to differing spectra (negative is small), but causing spread (positives broaden).
Example 2: NY660 and PE-Cy5
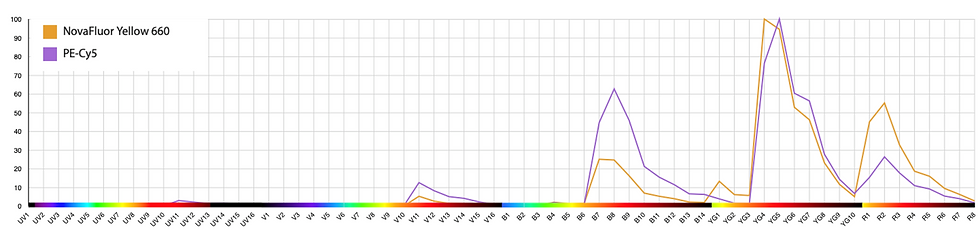

With this combination, we get spread from PE-Cy5 into NY660, but little the other way around. The negative is minimally distorted.
Example 2: NY610 and PE-Dazzle 594


This pairing causes massive negative distortion. Resolution is greatly impaired. There is also spread from PE-Dazzle 594 into NY610, with what looks to be less in the other direction (NY610 -> PE-Dazzle 594).
Also, I'll point out that this phenomenon isn't unique to spectral cytometers. We're more likely to encounter the issue on spectral cytometers because spectral cytometers generally allow us to better separate similar fluorophores, encouraging the use of combinations that create this issue. This problem also increases as panel size approaches the limits of the machine.
Here's an example of negative spreading on a conventional cytometer:

No BV750 With BV750

Impact of using the BV750 channel on the FACSymphony (Left: BV750 channel not included in the compensation matrix. Right: BV750 included). Note that we get an angled (distorted) negative in the upper right between BV750 and BV785. Thus, we get reduced resolution even on CD3+CD19- T cells versus BV785 (lower right).
So, spectral cytometers allow us to use practically any fluorophore that is excited by the lasers on our machine. How do we determine which combinations will cause a loss in resolution? You're probably heard that spectral unmixing allows us to use fluorophores with the same peak emission but different spectral profiles, such as touted examples with PE and dyes like Spark Yellow-Green 570, or combinations like APC and AF647. The similarity of the first two is 0.93, the second two, 0.9.
How true is this?
Here we’re looking at the normalized emission spectra for APC vs AF647:

Straight away we see that APC and AF647 don't actually share the same peak emission detector on the Aurora. These two are actually separable on a conventional cytometer with appropriate filters, and they work reasonably well as a pair on the Aurora.
Example of AF647 and APC together.

In contrast, PE and SYG570 peak in the same channel, YG1:

Differences in violet and blue excitation allows unmixing, but is it any good?
I’m going to use this pair as an example to show you why I think that using two fluorophores that peak in the same detector is almost never a good option. Personally, I recommend designing panels for spectral cytometers along the same principles as for conventional machines: using one fluorophore per detector. We just have more detectors to work with on spectral machines.
For this example, I’m using CD8 in PE and the macrophage marker F4/80 in SYG570, so there’s no co-expression and this is a best-case scenario. We can unmix these, but there is considerable distortion of the negative due to similarity of the fluorophores and the uncertainty that results. This negative spreading is independent of the how bright the staining is and causes a general loss of resolution. By this, I mean that the negative events are now reaching 10^4 on both the x and y axes. We can't resolve anything as definitely positive until almost 5x10^4.

Using very similar fluorophores causes a large loss in resolution.
Why is this happening? We have two bright fluorophores, so we have lots of signal. The positive events are located above 10^5.

Limited differences mean most values are uncertain.
However, most of the signal is indistinguishable. We can basically only separate based on these shaded areas in the violet and blue, which are not the majority of the signal.
We can make this look better by compressing the negative with the biexponential transform. We still have a lot of spread, but we can identify the populations.

Aesthetically more pleasing, but the separation is unchanged.
But, that’s only three colors: PE, SYG570 and fixable viability dye eFluor780. We’d like to include this in a high parameter panel, right? If we unmix twenty colors, the distortion of the negative increases. This is because we’ve been using several of the 64 detectors to better separate the PE and SYG570, and now we need to use some of those channels to detect and separate our additional fluorophores. What I’m showing here is still only stained with three colors; there are 17 empty unstained channels.

Not viable in medium-sized panels, even without fluorophore spread.
Using those channels adds more spread.

Pretty terrible in large panels.
New fluorophores are coming out all the time, and there are already decent combinations that allow for simultaneous detection at least 45 without relying on stacking two fluorophores in the same detector. There are some exceptions to this rule, and we'll look at one example in the next post.
Can anything be done to reduce negative distortion in existing data?
Sometimes, yes. Consider that the distortion arises from uncertainty in the unmixing algorithm--the computer can't figure out where to position the events accurately. If the single-color controls aren't good (few events, messy signatures, dim), the distortion may be larger than it would be otherwise. So, practically, you can go back and fix it by re-running the unmixing with better controls. In some cases, you may also get better results using a different unmixing algorithm (panelBuildeR offers some choices if you want to play around).
If, however, you're using very similar fluorophores, probably not much can be done at present. Personally, I believe there is scope for improvement in the unmixing math, particularly when it comes to using cell controls.
Further reading
To learn more about the phenomenon of negative spreading, check out these links:
Recent pre-print from Konecny et al. describing, among other things, an automated process for selecting fluorophores that will limit this distortion.
Seminar by Peter Mage (BD) on the math behind spectral unmixing and how this affects resolution.
Webinar by Robert Balderas (BD) that touches on how using similar fluorophores can lead to loss of resolution (registration required).
Cool article written by Mario Roederer showing in silico how compensation and spread are related. Credit to Florian Mair for the suggestion.



Comments